Instrument Parameter Optimization and Proteome Discoverer 2.1 Software Data Processing Method for TMT Quantitative Workflow in Q Exactive Series Instruments Xiaoyue Jiang1, Tabiwang Arrey2, Eugen Damoc2, Michaela Scigelova2, David Horn1, Rosa Viner1, and Andreas FR Huhmer1 Key words Research Objectives <br>The Thermo ScientificTM TMTsixplexTM and TMT10plexTM workflows were used on the Thermo ScientificTM Q ExactiveTM instrument platform to create optimal mass spectrometry data acquisition methods for relative quantitative studies of proteins. This article details the process of sample preparation, peptide separation, and mass spectrometry involved in this workflow, as well as the specific process for processing and analyzing data using the Thermo ScientificTM Proteome DiscovererTM software version 2.1. Background Introduction <br> Isobaric labels (such as Tandem Mass LabelTM (TMTTM) 1 or isobaric Tag for Relative and Absolute Quantification (iTRAQ®) 2) are very common in mass spectrometry based protein relative quantification methods. 3-6 Among them, the TTM-based multiple relative quantification method has many advantages, including shorter experimental periods, less fluctuations, higher sample throughput, and less loss of quantitative data between samples. The amino-tagged TMT10plex has the same chemical structure as the TMTsixplex reagent but the former provides more 13C and 15N isotopic combinations in the mass reporting region. For some combinations, the quality of a neutron differs from each other, and high-resolution analytical instruments are required to achieve tandem mass spectrometry acquisition. The Thermo ScientificTM TribridTM mass spectrometer family, including the OrbitrapTM FusionTM MS and Orbitrap Fusion LumosTM MS instruments, provides the Synchronous Master Ion Select (SPS) MS3 method for the most accurate TMT quantification in complex samples with high dynamic range. 7,8 and the Thermo ScientificTM Q ExactiveTM mass spectrometer family of instruments combines the high selectivity and high specificity of the quadrupole for parent ion selection with the high resolution and accurate mass (HRAM) detection capabilities of the Orbitrap mass analyzer. It is also a good hand for quantitative analysis of the same amount of different orders. The Thermo ScientificTM Q ExactiveTM Plus mass spectrometer is equipped with improved quadrupole technology (AQT) to optimize ion selection and transmission while improving the ability to quantify low abundance ions in a narrow isolation window. 9 The Thermo ScientificTM Q ExactiveTM HF Mass Spectrometer introduces the Ultra High Field Orbitrap Mass Analyzer, which delivers high performance detection at twice the scanning speed of the Q Exactive Plus MS for superior performance. 9 The QT-based quantitative analysis process based on the Q Exactive mass spectrometer not only maximizes peptide and protein identification results, but also provides relatively quantitative results for high quantitative accuracy for up to ten different samples simultaneously. The premise of this process is the need for good chromatographic separation, well-resolved mass spectral peaks, sufficient ion abundance, and the ability to acquire high quality MS2 fragmentation spectra at high scan rates. This application note provides detailed step-by-step instructions for TMT markers and their relative quantification strategies, including sample preparation, instrument setup, and processing and analysis of multiple quantitative data using the Thermo ScientificTM Proteome DiscovererTM 2.1 software. This paper discusses in detail the impact of key instrument parameters (resolving power, maximum injection time, automatic gain target value, collision energy and isolation window) on protein and peptide identification and quantitative results to ensure that users can successfully refer to this article in Q Exactive Establish a TMT quantitative workflow on a series of mass spectrometers. experimental method TMT Labeling <br>The E. coli digested product standard sample (Waters® Corp, Milford, MA) was labeled with the Thermo Scientific TMT reagent as follows: Liquid chromatography <br> chromatographic separation conditions are set forth in Table 1. The injection volume is equivalent to 1 μg of starting protein. Mass Spectrometers <br> The peptides eluted by liquid chromatography were analyzed by Q Exactive Plus and Q Exactive HF mass spectrometers in a data-dependent acquisition mode. See Table 2 for instrument parameter settings. 2-Piece Ostomy pouch MDK-BO-02 300ML
A one-piece colostomy bags, drainable pouching system designed to be opened at the bottom when emptying. These colostomy bag machine are most suitable for colostomies or ileostomies. Ostomy bag colostomy barrier is a standard wear skin barrier that is gentle to the skin and allows for frequent pouch removal. To close the pouch, use the curved, beige clamp. Soft, beige disposable colostomy bag pouch panels on body side help provide comfort.
The features are soft and flex,standard wear, skin barrier, flat. The Chassis made in 100% hydrocolloid No tape border. Cut-to-fit skin barrier. Ultra-clear odor-barrier pouch film. Curved, beige pouch clamp. With comfortable ware pouch panel body side only. Not made with natural rubber latex.
Ostomy urostomy colostomy bag care products consist of sodium carboxymethylcellulose, adhesives made from medical hot melt adhesives, and separator paper or separators. Nursing equipment for ileum, colon, rectum or urethral stoma,The coloplast colostomy bag comes into contact with intact skin and intestinal lumen,Non-sterile supply. Colostomy bag reusable for stoma washing, care and collection of excreta and skin care around the stoma.
Use disposable colostomy bag, make sure the skin around the stoma is clean and dry before use. Remove the bags for colostomi from the package and separator or paper. Reshape the coloplast colostomy bag into the desired shape and size to fit the skin around the stoma. Apply the colostomy convatec bag to the skin around the stoma,adjust the shape of the paste again,Gently press so that it is firmly flat on the skin. Attach the stoma colostomy bag undercarriage to the stoma. The colostomy bag care can be peeled off the skin, don`t reuse. These one-piece-colostomy bag or two-piece-colostomy-bag are non-sterile product and is valid for three years under the condition of meeting the storage conditions.
1-Piece Ostomy Pouch,One Piece Ostomy Bags,One Piece Ostomy Pouch,1 Piece Ostomy Bag Henan Maidingkang Medical Technology Co.,Ltd , https://www.mdkmedicales.com
1Thermo Fisher Scientic, San Jose, CA, USA; 2Thermo Fisher Scientic, Bremen, Germany
Q Exactive Plus, Q Exactive HF, TMT, Proteome Discoverer 2.1, Protein Identification, Relative Quantitation, Isobaric Labeling, Multiple Quantitation
1. Transfer the samples in the triethylamine carbonate (TEAB) buffer, TMT label, and dry powder from -20 °C to room temperature.
2. Add 500 μL of Dissolving Buffer (1 M TEAB) to 4.5 mL of ultrapure water to prepare a 100 mM TEAB solution.
3. Dissolve a single sample (up to 100 μg/TMT label) in 100 mM TEAB buffer to obtain a 1 μg/uL sample solution, vortex thoroughly and allow to stand for 10 minutes.
4. Add 41 μL of fresh LC-MS grade acetonitrile to each TMT vial and vortex thoroughly for 10 minutes.
5. Add no more than 100 μL of sample solution prepared in step 3 to each TMT vial, vortex and incubate for 1 hour.
6. After 1 hour of reaction, add 8 μL of 5% hydroxylamine (10% hydroxylamine stock solution provided in the kit diluted 10 times with water) to each vial to stop the reaction and incubate for 15 minutes.
7. Immediately after the end of the mark, mix the different samples in the desired ratio. In this study, the mixing ratio of TMTsixplex reagent (channel 126-131) was 20:10:1:1:10:20. The six selected TMT10plex reagents (channels 127C, 128N, 128C, 129N, 129C, 130N) were mixed in the same ratio.
8. Divide the prepared sample into small portions, dry and store at -80 °C.
9. Resuspend the prepared TMT-labeled sample in 0.1% TFA/5% ACN (v:v) solvent prior to LC-MS analysis; the resuspended TMT-labeled sample can be stored stably at 4 °C for one week. Avoid repeated freezing and thawing.
To investigate the accuracy of the quantitative analysis, the Thermo ScientificTM PierceTM HeLa Proteolytic Product Standard Sample (P/N 88328) was labeled as described above, with TMTsixplex channels 129–131 and TMT10plex channels 127N, 127C and 128N at 10:10, respectively: 10 Proportionally mixed and then added to the E. coli sample at a ratio of 1:1 per channel (33 μg HeLa hydrolysate + 67 μg E. coli digested product).
Data processing was performed using Proteome Discoverer software version 2.1 (PD 2.1). The software features a new and improved TMT quantitative analysis workflow with new features including: 1) a new user interface for adding reported ion isotope distributions; 2) the ability to resolve ion isotope impurities in TMT10plex reagents; Quantitative analysis of TMT based on signal-to-noise ratio (S/N value); 4) Quantitative analysis of proteins and peptides based on S/N values; 5) Normalization of peptide and protein abundance values Standardize new methods.
1. Go to Administration and select Maintain Quantification Methods. It provides a template for commonly used quantitative analysis methods.
3. The isotope distribution values ​​shall be added in accordance with the information provided in the Institute using the TMT Reagent Analysis Certificate (CoA). Information can be obtained by querying the lot number on the reagent bottle at http://?ICID=search-product.
4. Select/remove the report ion channel in the “active†column as needed for your research.
5. Save the newly created quantitative analysis method. See Figure 1 for the newly created research-specific quantitative analysis method.
2. Import the FASTA database
2. Use the Add function to import the existing .FASTA file on your local hard drive.
3. Another method is to use the Download function, type the taxonomy ID, and download the database file from the Protein Center. 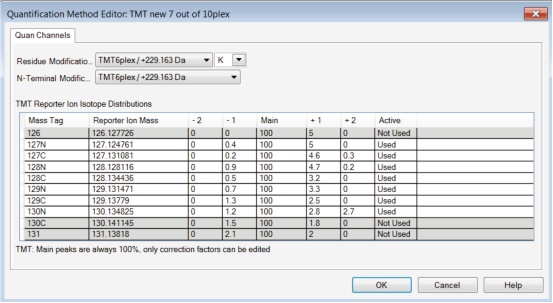
1. Select New Study and Analysis on the Proteome Discoverer software start page (Figure 2).
2. Define the Study Name and specify the Study Directory to save the file in a specific folder.
3. To perform the Processing Workflow, select the available template by the inverted triangle icon and select "ProcessingWF_QE active\PWF_QE_Reporter_Based_Quan_Seque tHT_Percolator.pdProcessingWF".
4. To make a Consensus Workflow, select "ConsensusWF CWF_Comprehensive_Enhanced_Annotation_Quan pdConsensusWF".
5. Select the Quantification Method created in the first section and select Control channel Selection.
7. Select OK and the data file will open in a new window.
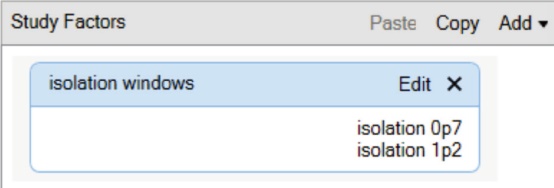
8. For complex studies involving parallel analysis, it is recommended to use the Study Factors management function for grouping, normalizing, and presenting results. To add Study Factors, select the Study Definition tab on the current study page and select Add to add factors for the category or number type. Type the factor name and all related factors (Figure 3). If you do not need to use the Study Factor, go directly to the workflow setting operation in Part IV.
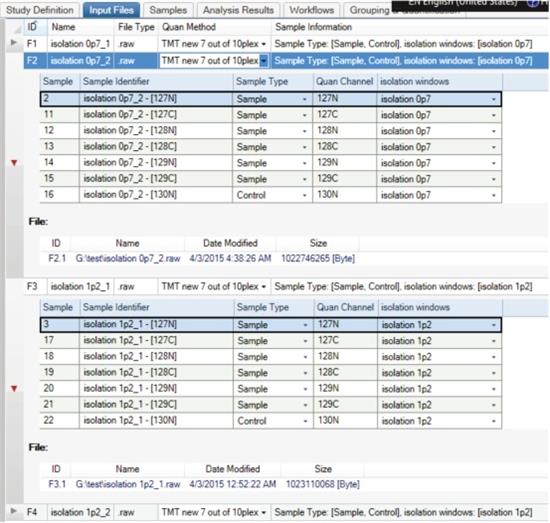
9. The imported original file should be displayed under the Input Files tab entry. You can expand the file details with the left triangle icon. Specify Sample Type and Study Factors for each file and report ion based on the experimental setup.
10. Review the Sample tab when you are done - all research factors should have been automatically updated. 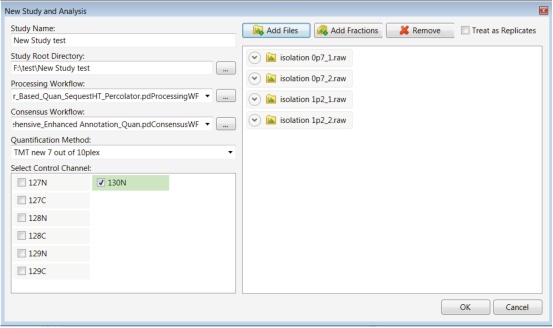
1. Under the SequestHT node of Processing Workflow, select Protein Database. Multiple protein databases can be selected simultaneously.
2. Under the SequestHT node of Processing Workflow, specify TMT as the static modification on the nitrogen and lysine residues of the peptide. (Note: both TMTsixplex and TMT10plex correspond to a 229.163 Da mass increase).
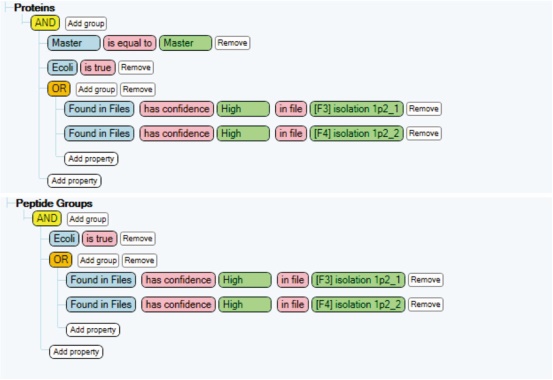
4. Open the consensus Workflow and select the Protein Marker node. Select the proteins that need to be indicated in the results. Since the sample is a mixture of E. coli and human protein, two types of markers are defined for subsequent analysis (E. coli and human) (Figure 5).
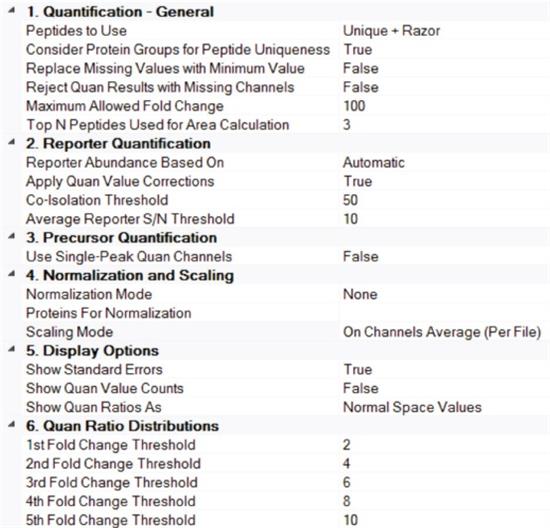
5. Enter the Peptide and Protein Quantifier node. Set Apply Quan Value Corrections to True, Co-Isolation Threshold to 50, and Average Reporter S/N Threshold to at least 10 (Figure 6). Scaling Mode uses On Channel Average (Per File), so that for each parallel analysis result, the abundance value of each channel will be scaled. See Reference 11 for more details on how parameters are set.
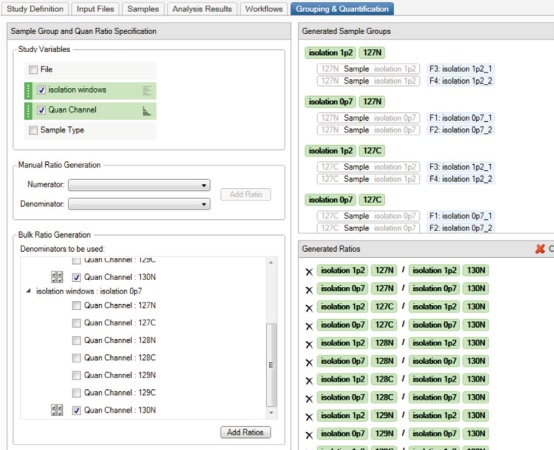
7. Go to the Grouping & Quantification entry and indicate the variables in the study. This setting determines how the quantitative analysis results are grouped and displayed. In this case, Quan Channel and isolation windows are selected, so the results of the parallel analysis are grouped and averaged by different isolation windows, making it easy to compare in the final report. The specific way to do this is to select these two variables and move the isolation windows above the Quan Channel by selecting the up and down arrows to the left of each variable. Note that the order of the variables affects how the grouping is done in Bulk Ratio Generation. Select the control group 130N Quan Channel as the Denominator for these two extraction windows, then select Add Ratios. Generated Ratios See Figure 7. See Reference 11 for more details on how to set up study variables.
8. Click Run in the upper right corner of Analysis to start the Processing and Consensus Workflow searches in the Job Queue. 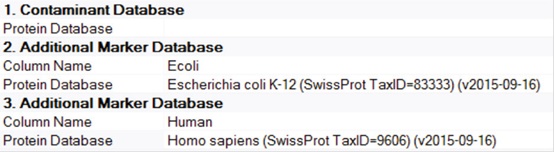
1. After the search is finished, highlight Consensus search and select Open Results. Or enter the Analysis Results entry, highlight the analysis line and select Open Results.
2. The number of groups of proteins and peptides is shown at the bottom of the screen.
3. For each protein/peptide group, the results of species assignment based on the protein marker nodes are also displayed (Figure 8).
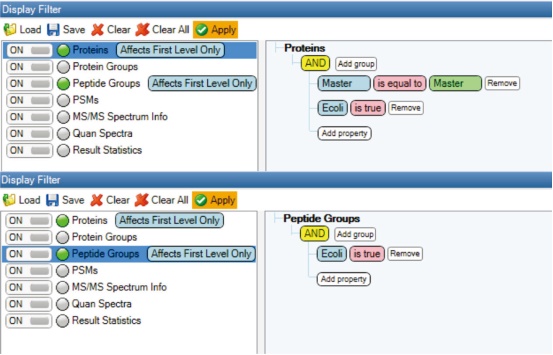
4. Go to the Toolbar View item and select Display Filter. All proteomes and peptide groups identified as E. coli were screened from all files (Figure 9).
5. To obtain the total number of identification results for each of the two study factors, screen using the conditions shown in Figure 10.
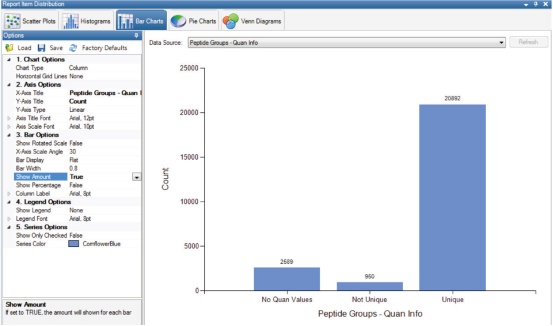
6. To get a quantifiable number of peptides, go to View and select Distribution Charts. Select Peptide Groups-Quan Info in Bar Charts to display peptide groups with and without Quan Values ​​(Figure 11). 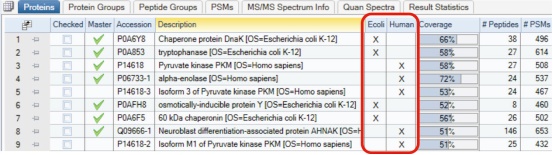
Careful setup of each mass spectrometry parameter is critical to achieving absolute sensitivity and accurate quantitative analysis based on TMT-labeled experiments. In general, longer gradients provide better chromatographic separation, less ion suppression, and less co-elution interference. Highly resolved MS1 and MS2 spectra are indispensable for accurate peptide and protein relative quantification results. The MS2 maximum injection time, AGC, NCE, and MS2 isolation window settings affect the quality and quantitative results of the MS/MS spectrum. Therefore, these parameters should be carefully selected while ensuring high acquisition rates of the instrument.
The chemical modification of the peptide affects its hydrophobicity, which changes its chromatographic retention behavior. Elution of TMT-labeled samples typically requires a higher proportion of the organic phase. Tables 3 and 4 provide recommended gradients for chromatographic separation of unlabeled and labeled cell lysate samples, respectively, for a total gradient time of 2 hours. 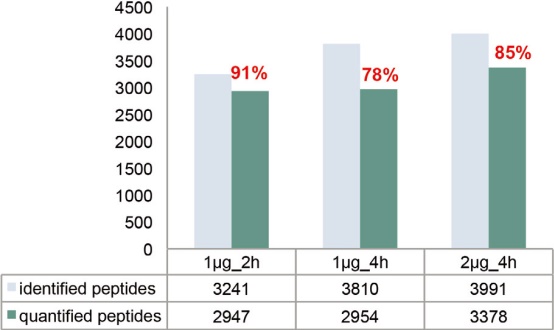
The MS2 maximum injection time needs to be adjusted based on injection volume, sample complexity, and elution gradient. Generally speaking, a longer injection time can accumulate more precursor ions, and the intensity of the reporter ions will increase accordingly, which is more conducive to the identification and quantitative analysis of the middle and low abundance peptides. As shown in Figure 16, the maximum injection time increased from 64 ms to 100 ms, which greatly increased the number of quantifiable proteins and peptides, and correspondingly achieved a higher quantitative analysis rate.
As mentioned earlier, longer elution gradients require longer injection times. On the other hand, the increase in injection time should not significantly affect the overall speed of the instrument, especially when analyzing complex samples. An ideal method is to set the maximum allowable injection time to match the FT conversion detection time, since this time is directly related to the instrument resolution setting. For Q Exactive Plus MS and Q Exactive HF MS, the 128 ms Orbitrap analyzer detection time corresponds to resolutions of 35,000 and 60,000, respectively, so the 100 ms injection time matches the detection time to ensure ion implantation and Orbitrap Efficient parallelism of detection. As shown in Figure 17, increasing the injection time is also beneficial to improve the accuracy of quantitative detection. 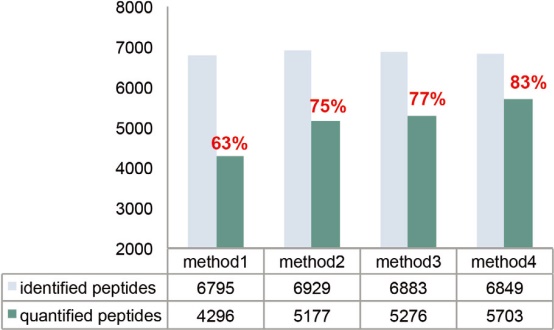
The automatic gain target value is another key parameter that needs to be set. The size determines the maximum number of ions that C trap can accumulate and enter the Orbitrap Analyzer for detection. Although the S/N and identification efficiencies increase with increasing ion number, the higher AGC target setting can cause ion accumulation or other space charge related problems. 13 Figures 16 and 17 compare the quantitative results of the AGC target values ​​set to 5e4 and 1e5 when the injection volume is 1 μg. When this parameter is set to 1e5, it is possible to achieve better quantification and precision on the basis of obtaining more quantifiable peptides without causing ion aggregation.
MS2 collision energy
Instruments of the Q Exactive type typically recommend a 2 Th isolation window to ensure adequate ion transport and sensitivity. However, expanding the isolation window will isolate the interfering ions from the parent ions, reducing the accuracy of the quantitative analysis of the reporter ions.
In addition, fragments of interfering ions may also contaminate the MS2 spectrum and result in reduced identification scores. Both Q Exactive Plus MS and Q Exactive HF MS are equipped with AQT, enabling efficient ion transfer in very narrow isolation window settings. We analyzed the E. coli samples at the isolation window settings of 0.7 and 1.2 Th, respectively, and the identification and quantification results were very close (Figure 18). 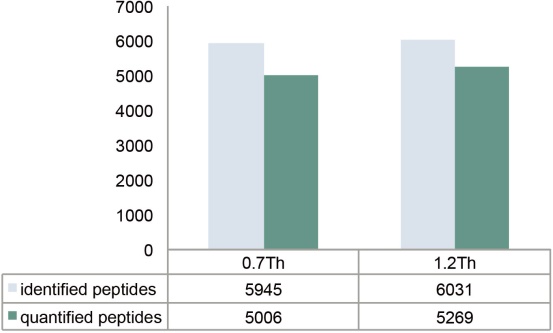
1) Use longer elution gradients, longer columns and higher loadings.
2) Adjust the MS2 resolution setting for different TMT tags.
3) Match the maximum injection time to the resolution to achieve an optimal mass spectrometry duty cycle.
4) Increase the AGC MS2 target based on the number of tags. The TMT10plex can be set to 1e5.
5) Improve NCE without excessive fragmentation of the peptide.
6) The quadrupole is a narrower parent ion isolation window than the conventional method, and is more suitable for the application of shotgun sequencing.
TMT markers increase sample throughput and support relative quantification of up to ten different biological samples, including cells, tissues and body fluids. This study evaluated the performance of Q Exactive Plus MS and Q Exactive HF MS instruments for protein/peptide identification efficiency and quantitative analysis accuracy using TMTsixplex and TMT10plex-labeled E. coli cell lysate samples.
Q Exactive Plus MS and Q Exactive HF MS are ideal for analyzing low to medium complexity samples. For high complexity samples, it is best to use pre-grading to improve the accuracy and accuracy of quantitative analysis. By comparing and optimizing multiple sets of different parameter settings, including MS2 resolution, maximum injection time, automatic gain target value, and isolation window, the highest identification efficiency and the most accurate quantitative analysis are achieved. At the same time, the paper also details the related settings of sample preparation and liquid chromatography, and how to use Proteome Discoverer software version 2.1 for data processing methods and processes.
1. Schäfer, T. et al. Anal. Chem. 2003, 75(8), 1895.
2. Ross, P. et al. Mol Cell Proteomics 2004, 3(12), 1154.
3. Savitski, M. et al. Science 2014, 346 (6205), 1255784.
4. Weekes, M. et al. Cell 2014, 157(6), 1460.
5. Rauniyar, N. et al. J Proteome Res. 2014, 13(12), 5293.
6. Christoforou, A. et al. Anal Bioanal Chem. 2012, 404(4), 1029.
7. Erickson, B. et al. Anal. Chem. 2015, 87(2), 1241.
8. Ting, L. et al. Nature Methods 2011, 8, 937.
9. Scheltema, R. et al. Mol Cell Proteomics 2014, 13(12), 3698.
10. McAlister, G. et al. Anal. Chem. 2012, 84(17), 7469.
11. Thermo Scientific Proteome Discoverer 2.1 User Guide
12. Viner, R. et al. Thermo Fisher Scientific Application Note 566.
13. Werner, T. et al. Anal. Chem. 2014, 86(7), 3594.
14. Arrey, T. et al. Thermo Scientific Poster Note 64412. 

400 650 5118 (support mobile phone users)
parameter
Setting
LC
Thermo ScientificTM EASY-nLCTM 1000
Mobile phase
A: 0.1% formic acid aqueous solution; B: 0.1% formic acid acetonitrile solution (Fisher Chemical)
Elution gradient
0-100 min 7–25% B; 100-120 min 25–60% B; 121 min 95% B, 95% 5 min
Flow rate
300 nL/min
Catch column
Thermo ScientificTM AcclaimTM PepMapTM 100 C18 LC Column, 75 μm x 2 cm, Thermo ScientificTM DionexTM nanoViperTM, C18, 3 μm, 100 Å (P/N 164705)
Separation column
Thermo ScientificTM EASY-SprayTM C18 LC Column, 75 μm x 50 cm, 2 μm, 100 Å (P/N ES803)
Table 2. Recommended Instrument Parameter Settings for Analysis of TMTsixplex and TMT10plex Labeled Samples with Q Exactive Plus and Q Exactive HF Instruments
Q Exactive Plus
Q Exactive HF
Full scan MS parameters
Resolving power (FWHM at m/z 200)
70,000
120,000
Automatic gain target value (AGC)
3e6
3e6
Maximum injection time (ms)
50
50
Scanning range (m/z)
375-1400
375-1400
MS2 parameters
Resolving power (FWHM at m/z 200)
35,000
30,000 (TMTsixplex)
60,000 (TMT10plex)
Automatic gain target value (AGC)
1e5
1e5
Maximum injection time (ms)
100
100
Isolated window (Th)
0.7 or 1.2
0.7 or 1.2
Normalized collision energy (NCE)
32
32
Loop count
10 or 15
10 or 15
Initial fixed mass ratio (First fixed mass)
100
100
Underfill ratio
2%
2%
Charge state recognition
2-7
2-7
Peptide match
Optimal
Optimal
Dynamic exclusion(s)
30
30
data processing
The key steps in setting up the data processing flow in the Proteome Discoverer 2.1 software are described in detail below:
1. Establish a research-specific quantitative analysis method <br>Before starting a new study, it is usually necessary to update the quantitative analysis method. The editing process is:
2. Select Add to select a template from an existing method. Each existing TMT quantitative analysis template contains a modification of the nitrogen end of the residue/peptide and an isotopic distribution of the reporter ion.
1. Open Maintain FASTA Files under the Administration entry.
Figure 1. Example of a user-defined research-specific TMT10plex quantitative analysis method.
3. Create a new study
6. Import the .raw file to be processed. The original data file generated by four parallel analyses using two different isolation window settings was imported in the example in Figure 2.
Figure 2. New research and analysis setup dialog interface.
Figure 3. Adding new research factors to the Study Factor management interface.
Figure 4. Study Factors and Quantitative Analysis Channels for a single sample quantitation.
4. Definition of workflow settings and quantitative ratios <br>First open the Processing Workflow under the workflows tab. The template already contains mass tolerances, reported ion quantitative analysis instructions, and common modifications. Only the following parameters need to be refined.
3. Under the Report Ion Quantitation node, verify that the MS level is MS2.
6. Drag .raw from the Input Files tab to the specified area under the Analysis entry on the right.
Figure 5. Settings at the Protein Marker node in Consensus Workflow for this study.
Figure 6. Parameter settings at the Peptide and Protein Quantifier node in Consensus Workflow.
Figure 7. The contents of the Grouping & Quantification label when performing a scale calculation.
5. Protein and peptide identification and quantitative results analysis
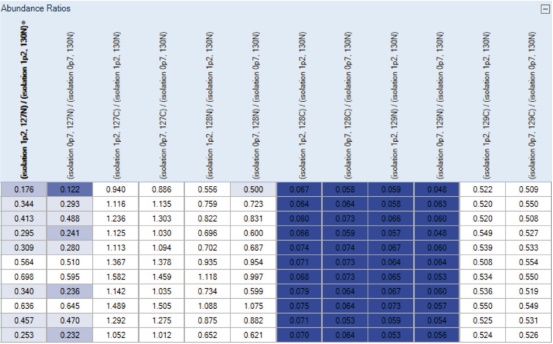
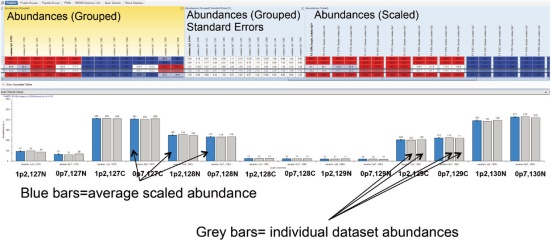
7. As designed in the Grouping & Quantification tab, the quantitative ratios of the two parallel analyses under each condition are grouped and averaged, as shown in the Abundance Ratios column (Figure 12). In addition, Grouped and Scaled Abundances, Standard Error between parallel analyses, and Scaled Abundances for each parallel analysis data set are displayed (Figure 13). These features make it easy to compare the results of quantitative analysis under different conditions.
Figure 8. Proteins identified by source of species.
Figure 9. E. coli proteome (top panel) and peptide group (bottom panel) identification results sieve.
Figure 10. Filters for the identification of proteome (top panel) and peptide group (bottom panel) using each of the study factors in this study.
Figure 11. Overview of the results of quantitative analysis of the screened peptide groups.
Figure 12. Abundance Ratios column in the results. The ratios are sorted by Isolation Windows and Quan Channel, respectively. Proteins that are up- or down-proportioned are highlighted in different colors. The darker the color, the more significant the change.
Figure 13. Graphical description of Abundances (Grouped), Standard Errors, and scaled Abundances for each data set.
Results and discussion
Liquid chromatography
Table 3. LC gradients for unlabeled samples.
time
duration
Flow rate
%B
0
N/A
300
2
5
5
300
2
105
100
300
20
125
20
300
32
126
1
300
95
134
8
300
95
Table 4. LC gradients for TMT labeled samples.
time
duration
Flow rate
%B
0
N/A
300
5
3
3
300
5
5
2
300
7
105
100
300
25
125
20
300
60
126
1
300
95
134
8
300
95
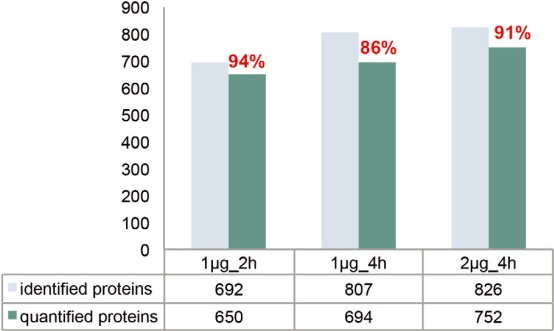
As the complexity of the sample increases, the elution gradient should be lengthened accordingly (eg, 4 hours) to ensure better chromatographic separation. However, lengthening the elution gradient inevitably leads to broadening of the chromatographic peak and a decrease in the parent ion S/N. Therefore, consideration should be given to increasing the injection volume or increasing the maximum injection time limit. The final indicator used to evaluate the experimental effect should be the number of quantifiable peptides, and the so-called quantifiable peptide refers to those fragments in which the fragment spectrum contains all predicted reporter ion peaks. As shown in Figure 14A, for the 1 μg injection volume, the elution gradient was increased from 2 hours to 4 hours, and the number of identified peptides increased from 3,241 to 3,810. The results of the identification clearly benefited from an improvement in the separation effect, as the increased sensitivity of the detection of low abundance peptides resulted in a corresponding increase in protein sequence coverage. On the other hand, the number of quantifiable peptides did not increase because the maximum injection time setting was the same as the 2-hour gradient (Table 2). If the injection volume is increased to 2 μg while lengthening the elution gradient, more than 400 quantifiable peptides are added, increasing the proportion of quantifiable/identifiable peptides to 85%. Similarly, the number of quantifiable proteomes also increased with increasing chromatographic gradient time and injection volume (Figure 14B). This result indicates that for high dynamic range samples (20:1), the best match between chromatographic separation conditions, injection volume and maximum injection time is achieved to obtain reliable quantitative results.
MS1 parameter
The TMT mark is a true isobaric label for the parent ion, so the MS1 resolution is set exactly the same as the unlabeled sample: For Q Exactive Plus MS and Q Exactive HF MS, the resolution is 70,000 respectively. And 120,000. Such a setting is sufficient to resolve most of the co-eluting material without significantly affecting the data-dependent scan mode cycle time. The recommended AGC target is 3e6, which helps to improve the dynamic range of the full sweep.
MS2 resolution
Insufficient resolution of MS2 can cause background or impurity ion signals to be confused with reported ion signals, affecting the evaluation of S/N. For TMTsixplex-labeled samples, a resolution of 30,000 ensures baseline separation of the reported ions from the isotope peaks of similarly reported ions. 12
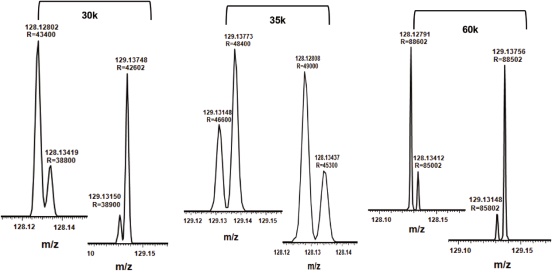
For the TMT10plex reagent, the mass difference between the 13C and 15N isotope is only 6 mDa. Shown in Figure 15 are the reporter ion regions of peptides labeled with 4 channels (128N, 128C, 129N, 129C) at different ratios. The resolution setting of 30,000 is not sufficient to distinguish between the N and C isotope isomers. For TMT10plex-labeled samples, the appropriate Q Exactive Plus MS and Q Exactive HF MS resolution settings should be 35,000 and 60,000, respectively.
Figure 14. Analysis of the amount of E. coli source peptides and proteins that can be identified and quantified after increasing the injection volume and gradient elution time for TMTsixplex-labeled E. coli and Hela mixed samples.
Figure 15. TMT10plex labeled peptides report ion region detail maps with resolution settings of 30,000, 35,000, and 60,000, respectively. Please note that only partial separation is obtained at 30,000 resolution.
MS2 maximum injection time
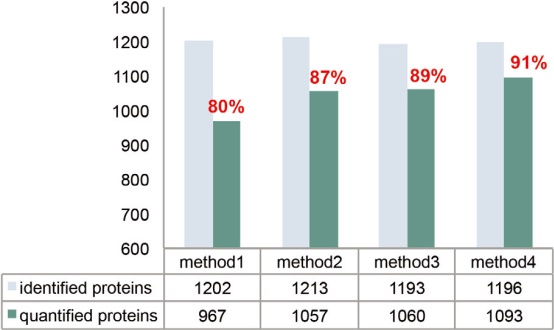
Figure 16. The number of peptides and proteomes that can be identified and quantifiable in pure E. coli samples using different instrument settings. *See Table 5 for details on method settings.
Table 5. Detailed parameters for each method compared in Figure 16.
MS2 IT
MS2 AGC
MS2 NCE
Method 1
64 ms
5e4
30
Method 2
100 ms
5e4
30
Method 3
100 ms
1e5
30
Method 4
100 ms
1e5
32
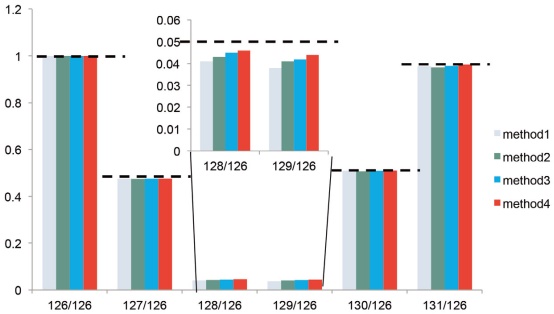
Figure 17. Ratio of TMT-labeled pure E. coli. The expected ratio is indicated by a dotted line. Please note that for the 128/126 and 129/126 ratios, the quantitative analysis of Method 4 is the most accurate.
MS2 automatic gain target value (AGC)
NCE is a dimensionless number roughly equivalent to the collision energy (in eV) of a reference ion with a mass of 500 and a charge of 1. The actual energy used is automatically calculated in real time based on the mass and charge number of the selected parent ion. The commonly used NCE settings (eg, NCE 27–28) are capable of generating enough fragment ions for peptide identification, but it has proven to be insufficient to dissociate and accurately quantify TMT reporter ions. However, if the collision energy is too large, it will lead to excessive fragmentation of the peptide and reduce the identification efficiency. 14 Studies have found that increasing collision energy from NCE 30 to NCE 32 significantly increases the number of quantifiable proteins without negatively impacting the efficiency of the assay (Figure 16). In addition, high collision energy also improves the accuracy of quantitative analysis (Figure 17). Therefore, we recommend using a collision energy of 4-6 NCE higher for TMT-labeled samples than for unlabeled samples.
MS2 isolation window
Figure 18. Number of identifiable and quantifiable peptide groups under different isolation window settings. The displayed data is the combined result of two parallel analyses of the E. coli sample.
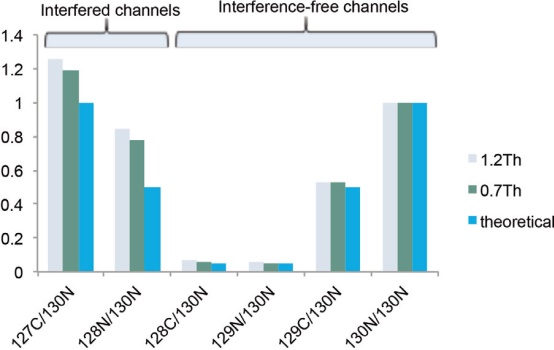
To evaluate the isolation window for complex samples, pure E.coli samples (127C:128N:128C:129N:129C:130N mixed in a ratio of 20:10:1:1:10:20) and equivalent HeLa enzymatic samples (Channels 127N, 127C, and 128N are mixed in a 1:1:1 ratio) Mix as background. The results showed that the quantitative ratios of the E. coli channels 127C and 128N affected by the interference of HeLa enzymatic products were significantly different from the theoretical values ​​and other non-interfering channels (Fig. 19). A narrower isolation window (0.7 Th) reduces co-extraction interference with a slightly better accuracy than a wider window (1.2 Th). For highly complex samples, we recommend the SPS MS3 method in the Orbitrap Fusion MS and Orbitrap Fusion Lumos MS instruments for accurate quantitative analysis at high dynamic range. In addition, sample sizing techniques (eg, Thermo ScientificTM PierceTM High pH Reverse Peptide Separation Kit, P/N 84868) can be used to reduce sample complexity.
In theory, a smaller isolation window (0.4 Th) can be used to improve the quantitative analysis accuracy of Q Exactive Plus MS or Q Exactive HF MS, but its quantitative sensitivity will also be affected.
Figure 19. Quantitative analysis accuracy for different isolation windows, expressed as the median of the quantitative ratio of quantifiable peptide groups.
In general, to perform a successful TMT experiment on any Q Exactive Series mass spectrometer, you need to carefully select the relevant parameters based on the characteristics of the sample. To achieve high sensitivity and high precision quantitative analysis goals, it is recommended to consider the following factors:
in conclusion
references
Orbitrap Group Club Thermo Fishery Mass Spectrometry Application Technology Group
National service hotline